Publications
2019
Zhao Y*, Mehta M*, Walton A*, Talsania K*, Levin Y, Shetty J, Gillanders EM, Tran B, Carrick DM. Robustness of RNA sequencing on older formalin-fixedparaffin-embedded tissue from high-grade ovarian serous adenocarcinomas. PLoS 2019 May 6;14(5): e0216050.
Levin Y†, Talsania K†, Tran B, Shetty J*, Zhao Y*, Mehta M*. Optimization for sequencing and analysis of degraded FFPE-RNA samples. JoVE, In Press. († Co-first authors; * Co-corresponding authors)
Magen A, Nie J, Ciucci T, Tamoutounour S, Zhao Y, Mehta M, Tran B, McGavern DB, Hannenhalli S, Bosselut R. Single-Cell Profiling Defines Transcriptomic Signatures Specific to Tumor-Reactive versus Virus-Responsive CD4+T Cells. Cell Reports, 2019, 29(10): 3019-3032.e6
The Somatic Mutation Working Group of the SEQC-II consortium, Xiao W, Kusko R, Ren L, Fang F, Shen T, Talsania K, Kriga Y, Shetty J, Tran B, Zhao Y, et al. Towards best practice in cancer mutation detection with whole-genome and whole-exome sequencing. Nat Biotechnol, 2019. Accepted
Ma L, Hernandez M, Zhao Y, Mehta M, Tran B, Kelly M, Rae Z, Hernandez J, Davis J, Martin S, Kleiner D, Hewitt S, Ylaya K, Wood B, Greten T, Wang X. Tumor Cell Biodiversity Drives Microenvironmental Reprogramming in Liver Cancer. Cancer Cell. 2019 Oct 03.
Jiao X, Sui H, Lyons C, Tran B, Sherman BT, Imamichi T. Complete Genome Sequence of Herpes Simplex Virus 1 Strain McKrae. Microbiol Resour Announc. 2019 Sep 26;8(39).
Jiao X, Sui H, Lyons C, Tran B, Sherman BT, Imamichi T. Complete Genome Sequence of Herpes Simplex Virus 1 Strain MacIntyre. Microbiol Resour Announc. 2019 Sep 12;8(37).
Vacchio MS, Ciucci T, Gao Y, Watanabe M, Balmaceno-Criss M, McGinty MT, Huang A, Xiao Q, McConkey C, Zhao Y, Shetty J, Tran B, Pepper M, Vahedi G, Jenkins MK, McGavern DB, Bosselut R. A Thpok-Directed Transcriptional Circuitry Promotes Bcl6 and Maf Expression to Orchestrate T Follicular Helper Differentiation. Immunity. 2019 Sep 17;51(3):465-478.e6. Epub 2019 Aug 15.
Talsania K, Mehta M, Raley C, Kriga Y, Gowda S, Grose C, Drew M, Roberts V, Tai Cheng K, Burkett S, Oeser S, Stephens R, Soppet D, Chen X, Kumar P, German O, Smirnova T, Hautman C, Shetty J, Tran B, Zhao Y, & Esposito D. Genome Assembly and Annotation of the Trichoplusia ni Tni-FNL Insect Cell Line Enabled by Long-Read Technologies. Gene, 2019, 10 (2). pii: E79.
Ciucci T, Vacchio MS, Gao Y, Ardori FT, Candia J, Mehta M, Zhao Y, Tran B, Tessarollo L, McGavern D, & Bosselut R. Emergence and functional fitness of memory CD4+ T cells require the transcription factor Thpok. Immunity, 2019, 50(1): 91-105.e4.
2018
Zheng H, Pomyen Y, Hernandez MO, Li C, Livak F, Tang W, Dang H, Greten T, Zhao Y, Mehta M, Levin Y, Shetty J, Tran B, Budhu A, and Wang XW. Single cell analysis reveals cancer stem cell heterogeneities in hepatocellular carcinoma. Hepatology, 2018, 68(1): 127-140.
Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, Roulland S, Kasbekar M, Young RM, Shaffer AL, Hodson DJ, Xiao W, Yu X, Yang Y, Zhao H, Xu W, Liu X, Zhou B, Du W, Chan WC, Jaffe ES, Gascoyne RD, Connors JM, Campo E, Lopez-Guillermo A, Rosenwald A, Ott G, Delabie J, Rimsza LM, Tay Kuang Wei K, Zelenetz AD, Leonard JP, Bartlett NL, Tran B, Shetty J, Zhao Y, Soppet DR, Pittaluga S, Wilson WH, Staudt LM. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med. 2018 Apr 12;378(15):1396-1407.
Miller ME, Zhang Y, Omidvar V, Sperschneider J, Schwessinger B, Raley C, Palmer JM, Garnica D, Upadhyaya N, Rathjen J, Taylor JM, Park RF, Dodds PN, Hirsch CD, Kianian SF, Figueroa M:De Novo assembly and phasing of dikaryotic genomes from two isolates of puccinia coronata f. sp. avenae, the causal agent of oat crown rust. MBio, 9(1):2018.
Greer YE, Porat-Shliom N, Nagashima K, Stuelten C, Crooks D, Koparde VN, Gilbert SF, Islam C, Ubaldini A, Ji Y, Gattinoni L, Soheilian F, Wang X, Hafner M, Shetty J, Tran B, Jailwala P, Cam M, Lang M, Voeller D, Reinhold WC, Rajapakse V, Pommier Y, Weigert R, Linehan WM, Lipkowitz S. ONC201 kills breast cancer cells in vitro by targeting mitochondria. Oncotarget. 2018 Apr 6;9(26):18454-18479.
Cramer SD, Hixon JA, Andrews C, Porter RJ, Rodrigues GOL, Wu X, Back T, Czarra K, Michael H, Cam M, Chen J, Esposito D, Senkevitch E, Negi V, Aplan PD, Li W, Durum SK. Mutant IL-7Rα and mutant NRas are sufficient to induce murine T cell acute lymphoblastic leukemia. Leukemia. 2018 Aug;32(8):1795-1882.
2017
Shukla A, Zhu J, Kim SY, Hager G, Ruan Y and Hunter KW (2017) Identification of a core inherited metastatic susceptibility network by integrated epigenetic, genetic and chromosomal interaction analysis. Manuscript in preparation
Carpenter AC, Wohlfert E, Chopp LB, Vacchio MS, Nie J, Zhao Y, Shetty J, Xiao Q, Deng C, Tran B, Cam M, Gaida MM, Belkaid Y, Bosselut R. Control of Regulatory T Cell Differentiation by the Transcription Factors Thpok and LRF. J Immunol. 2017 Sep 1;199(5): 1716-1728.
2016
Hodson DJ, Shaffer AL, Xiao W, Wright GW, Schmitz R, Phelan JD, Yang Y, Webster DE, Rui L, Kohlhammer H, Nakagawa M, Waldmann TA, Staudt LM. Regulation of normal B cell differentiation and malignant B cell survival by OCT2. Proc Natl Acad Sci 2016 113:E2039-E2046.
Thompson, Bethtrice; Varticovski, Lyuba; Baek, Songjoon; et al. Hager GL. Genome-Wide Chromatin Landscape Transitions Identify Novel Pathways in Early Commitment to Osteoblast Differentiation. PLOS ONE Volume: 11 Issue: 2
Yang Y, Kelly P, Shaffer AL, Schmitz R, Liu X, Huang DW, Webster D, Young RM, Yoo H, Nakagawa M, Ceribelli M, Wright GW, Yang Y, Zhao H, Yu X, Xu W, Chan WC, Jaffe ES, Gascoyne RD, Campo E, Rosenwald A, Ott G, Delabie J, Rimsza L, Staudt LM. Targeting non-proteolytic protein ubiquitination for the treatment of diffuse large B cell lymphoma. Cancer Cell 2016 29:494-507.
Kuschal C, Botta E, Orioli D, Digiovanna JJ, Seneca S, Keymolen K, Tamura D, Heller E, Khan SG, Caligiuri G, Lanzafame M, Nardo T, Ricotti R, Peverali FA, Stephens R, Zhao Y, Lehmann AR, Baranello L, Levens D, Kraemer KH, Stefanini M. GTF2E2 Mutations Destabilize the General Transcription Factor Complex TFIIE in Individuals with DNA Repair-Proficient Trichothiodystrophy. Am J Hum Genet. 2016 Apr 7;98(4):627-42.
Liang M, Raley C, Zheng X, Kutty G, Gogineni E, Sherman BT, Sun Q, Chen X, Skelly T, Jones K, Stephens R, Zhou B, Lau W, Johnson C, Imamichi T, Jiang M, Dewar R, Lempicki RA, Tran B, Kovacs JA, Huang DW. Distinguishing highly similar gene isoforms with a clustering-based bioinformatics analysis of PacBio single-molecule long reads. BioData Min. 2016 Apr 5; 9:13.
Huang DW, Raley C, Jiang MK, Zheng X, Liang D, Rehman MT, Highbarger HC, Jiao X, Sherman B, Ma L, Chen X, Skelly T, Troyer J, Stephens R, Imamichi T, Pau A, Lempicki RA, Tran B, Nissley D, Lane HC, Dewar RL. Towards Better Precision Medicine: PacBio Single-Molecule Long Reads Resolve the Interpretation of HIV Drug Resistant Mutation Profiles at Explicit Quasispecies (Haplotype) Level. J Data Mining Genomics Proteomics. 2016 Jan;7(1). pii: 182. Epub 2015 Nov 8.
Ma L, Chen Z, Huang da W, Kutty G, Ishihara M, Wang H, Abouelleil A, Bishop L, Davey E, Deng R, Deng X, Fan L, Fantoni G, Fitzgerald M, Gogineni E, Goldberg JM, Handley G, Hu X, Huber C, Jiao X, Jones K, Levin JZ, Liu Y, Macdonald P, Melnikov A, Raley C, Sassi M, Sherman BT, Song X, Sykes S, Tran B, Walsh L, Xia Y, Yang J, Young S, Zeng Q, Zheng X, Stephens R, Nusbaum C, Birren BW, Azadi P, Lempicki RA, Cuomo CA, Kovacs JA. Genome analysis of three Pneumocystis species reveals adaptation mechanisms to life exclusively in mammalian hosts. Nat Commun. 2016 Feb 22;7:10740.
Rui L, Drennan AC, Ceribelli M, Zhu F, Wright GW, Xiao W, Grindle KM, Lu L, Hodson DJ, Zhao H, Xu W, Yang Y, Staudt LM. Epigenetic gene regulation by Janus kinase 1 in diffuse large B cell lymphoma. Proc Natl Acad Sci, in press, 2016.
Smith OK, Kim RG, Fu H, Martin M, Utani K, Zhang Y, Marks AB, Lalande M, Chamberlaine S, Libbrecht MW, Bouhassira EE, Ryan MC, Noble WC, Aladjem MI. Distinct Epigenetic Features of Differentiation-Regulated Replication Origins. Epigenetics and Chromatin 9:18. 2016.
Zhang Y, Huang L, Fu H, Smith OK, Lin CM, Utani K, Rao M, Reinhold WC, Redon CE, Ryan M, Kim RG, You Y, Hanna H, Boisclair Y, Long Q, Aladjem MI. A Replicator-Specific Binding Protein Essential For Site-Specific Initiation of DNA Replication in Mammalian Cells. Nat. Commun. 7:11748. 2016.
Ceribelli M, Hou EZ, Kelly PN, Huang DW, Ganapathi K, Evbuomwan MO, Pittaluga S, Shaffer AL, Wright G, Marcucci G, Forman SJ, Xiao W, Guha R, Zhang X, Ferrer M, Chaperot L, Plumas L, Jaffe ES, Thomas CJ, Reizis B, Staudt LM. A druggable TCF4- and BRD4-dependent transcriptional network sustains malignancy in blastic plasmacytoid dendritic cell neoplasm. Cancer Cell 2016, in press.
Zhang M, Lykke-Andersen S, Zhu B, Xiao W, Hoskins JW, Jermusyk A, Zhang X, Rost L, Collins I, Jia J, Parikh H, Zhang T, Song L, Zhu B, Zhou W, Matters GL, Kurtz RC, Yeager M, Jensen TH, Brown KM, Bamlet WR, TCGA Research Network, Chanock S, Chatterjee N, Wolpin BM, Smith J, Olson SH, Petersen GM, Shi J, Amundadottir LT. Characterizing cis-regulatory variation in the transcriptome of histologically normal and tumor-derived pancreatic tissues. 2016: Gut
Doran AG, Wong K, Flint J, Adams DJ, Hunter KW* and Keane TM* (2016) Deep genome sequencing and variation analysis of 13 inbred mouse strains find novel missense mutations in essential DNA repair pathway genes. Genome Biology, 17:167.
Zhang S, Zhu I, Deng T, Furusawa T, Rochman M, Vacchio MS, Bosselut R, Yamane A, Casellas R, Landsman D, Bustin M. HMGN proteins modulate chromatin regulatory sites and gene expression during activation of naïve B cells. Nucleic Acids Res. 2016 Sep 6;44(15):7144-58.
Deng T, Zhu ZI, Zhang S, Postnikov Y, Huang D, Horsch M, Furusawa T, Beckers J, Rozman J, Klingenspor M, Amarie O, Graw J, Rathkolb B, Wolf E, Adler T, Busch DH, Gailus-Durner V, Fuchs H, Hrabě de Angelis M, van der Velde A, Tessarollo L, Ovcherenko I, Landsman D, Bustin M. Functional compensation among HMGN variants modulates the DNase I hypersensitive sites at enhancers. Genome Res. 2015 Sep;25(9):1295-308.
Deng T, Zhu ZI, Zhang S, Leng F, Cherukuri S, Hansen L, Mariño-Ramírez L, Meshorer E, Landsman D, Bustin M. HMGN1 modulates nucleosome occupancy and DNase I hypersensitivity at the CpG island promoters of embryonic stem cells. Mol Cell Biol. 2013 Aug;33(16):3377-89.
Bai L, Yang H, Hu Y, Shukla, A, Ha, N-H, Doran A, Faraji F, Goldberger N, Lee M, Keane T and Hunter KW. (2016) An integrated genome-wide systems genetics screen for breast cancer susceptibility genes. PLoS Genetics.
Ha N-H, Long J, Cai Q, Shu X-O and Hunter KW. The circadian rhythm gene Arntl2 is a metastasis susceptibility gene for estrogen receptor-negative breast cancer. PLoS Genetics, 12(9) e1006267. The article highlighted by the journal (Siracusa and Bussard, PLoS Genetics 12(9) e1006299).
Kim J, Sturgill D, Tran AD, Sinclair DA, Oberdoerffer P. Controlled DNA double-strand break induction in mice reveals post-damage transcriptome stability. Nucleic Acids Res. 2016 Apr 20;44(7):e64.
Khurana S, Kruhlak MJ, Kim J, Tran AD, Liu J, Nyswaner K, Shi L, Jailwala P, Sung MH, Hakim O, Oberdoerffer P. A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Nucleic Acids Res. 2016 Apr 20;44(7):e64.
2015
Young RM, Wu T, Schmitz T, Dawood M, Xiao W, Phelan JD, Xu W, Menard L, Meffre E, Chan WC, Jaffe ES, Gascoyne RD, Campo E, Rosenwald A, Ott G, Delabie J, Rimsza L, Staudt LM. Survival of human lymphoma cells requires B cell receptor engagement by self-antigens. Proc Natl Acad Sci 2015 112:13447-54.
Manna S, Kim JK, Baugé C, Cam M, Zhao Y, Shetty J, Vacchio MS, Castro E, Tran B, Tessarollo L, Bosselut R. Histone H3 Lysine 27 demethylases Jmjd3 and Utx are required for T-cell differentiation. Nat Commun. 2015;6:8152
Miles, George; Zhao, Yongmei; Levin, Yelena; et al. Multiplex Tissue and Clinical Proteomics By Next-Generation Sequencing Conference: 104th Annual Meeting of the United-States-and-Canadian-Academy-of-Pathology Location: Boston, MA Date: MAR 21-27, 2015
Fu H, Martin MM, Regairaz M, Huang L, You Y, Lin CM, Ryan M, Kim R, Shimura T, Pommier Y, Aladjem MI. The DNA repair endonuclease Mus81 facilitates fast DNA replication in the absence of exogenous damage. Nature Communications 6:6746. 2015.
Bartholdy B, Mukhopadhyay R, Lajugie J, Aladjem MI, Bouhassira EE. Allele-specific analysis of DNA replication origins in mammalian cells. Nat Commun.6:7051. 2015.
2014
Schmitz R, Ceribelli M, Pitaluga S, Wright G, and Staudt LM. Oncogenic mechanisms in Burkitt lymphoma. Cold Spring Harb Perspect Med. 2014 4:1-13.
Yang Y, Schmitz R, Mitala J, Whiting A, Xiao W, Ceribelli M, Wright G, Zhao H, Yang Y, Xu W, Rosenwald A, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Wiestner A, Kruhlak MJ, Iwai K, Bernal F, Staudt LM. Essential role of the linear ubiquitin chain assembly complex in lymphoma revealed by rare germline polymorphisms. Cancer Discovery 2014 4:480-93.
Yudkin D, Hayward B, Aladjem MI, Kumari D, Usdin K. Chromosome fragility and the abnormal replication of the FMR1 locus in Fragile X syndrome. Hum Mol Genet, 23:2940-52. 2014.
Mukhopadhyay R, Lajugie J, Fourel N, Selzer A, Schizas M, Bartholdy B, Mar J, Lin CM, Martin MM, Ryan M, Aladjem MI, Bouhassira EE. Allele-specific genome-wide profiling in human primary erythroblasts reveals replication program organization. PLoS Genetics 10(5): e1004319. 2014.
Hoskins JW, Jia J, Flandez M, Parikh H, Xiao W, Collins I, Emmanuel MA, Ibrahim A, Powell J, Zhang L, Malats N, Bamlet WR, Petersen GM, Real FX, Amundadottir LT. Transcriptome analysis of pancreatic cancer reveals a tumor suppressor function for HNF1A. Carcinogenesis 2014; 35(12): 2670-2678.
Yi, Ming; Zhao, Yongmei; Jia, Li; et al. Performance comparison of SNP detection tools with Illumina exome sequencing data-an assessment using both family pedigree information and sample-matched SNP array data. NAR Volume: 42. Issue: 12 Article Number: e101
Muppidi JR, Schmitz R, Green JA, Xiao W, Larsen AB, Braun SE, An J, Xu Y, Rosenwald A, Ott G, Gascoyne RD, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Vaidehi N, Staudt LM*, Cyster JG*. Loss of signaling via Gα13 in germinal center B cell-derived lymphoma. Nature 2014 516: 254-8.
Ceribelli M, Kelly P, Shaffer AL, Wright G, Yang Y, Mathews-Griner LA, Guha R, Shinn P, Keller JM, Liu D, Patel PR, Ferrer M, Joshi S, Nerle S, Sandy P, Normant E, Thomas CJ, Staudt LM. Blockade of oncogenic IkB kinase activity in ABC DLBCL by small molecule BET protein inhibitors. Proc Natl Acad Sci 2014 111:11365-70.
Nakagawa M, Schmitz R, Xiao W, Goldman CK, Xu W, Yang Y, Yu X, Waldmann TA, Staudt LM. Gain-of-function CCR4 mutations in adult T-cell leukemia/lymphoma. J Exp Med 2014 211:2497-2505.
2013
Xiao W, Tran B, Staudt LM, Schmitz R. High-throughput RNA sequencing in B-cell lymphomas. Methods Mol Biol 2013 971:295-312.
Jia J, Parikh H, Xiao W, Hoskins JW, Pflicke H, Liu X, Collins I, Zhou W, Wang Z, Powell J, Thorgeirsson SS, Rudloff U, Petersen GM, Amundadottir LT. An integrated transcriptome and epigenome analysis identifies a novel candidate gene for pancreatic cancer. BMC Med Genomics 2013; 6:33.
Fu YP, Kohaar I, Rothman N, Earl J, Figueroa JD, Ye Y, Malats N, Tang W, Liu L, Garcia-Closas M, Muchmore B, Chatterjee N, Tarway M, Kogevinas M, Porter-Gill P, Baris D, Mumy A, Albanes D, Purdue MP, Hutchinson A, Carrato A, Tardón A, Serra C, García-Closas R, Lloreta J, Johnson A, Schwenn M, Karagas MR, Schned A, Diver WR, Gapstur SM, Thun MJ, Virtamo J, Chanock SJ, Fraumeni JF Jr, Silverman DT, Wu X, Real FX, Prokunina-Olsson L. Common genetic variants in the PSCA gene influence gene expression and bladder cancer risk. Proc Natl Acad Sci U S A. 2012 Mar 27;109(13):4974-9.
Swaminathan, Sanjay; Hu, Xiaojun; Zheng, Xin; et al. Interleukin-27 treated human macrophages induce the expression of novel microRNAs which may mediate anti-viral properties. BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS Volume: 434 Issue: 2 Pages: 228-234.
Fu H, Maunakea AK, Martin MM, Huang L, Zhang Y, Ryan M, Kim R, Lin CM, Zhao K, Aladjem MI. Methylation of histone H3 on lysine 79 associates with a group of replication origins and helps limit DNA replication once per cell cycle. PLoS Genet. 9:e1003542. 2013.
Collaborative Publications
2012
Snow AL, Xiao W, Stinson JR, Lu W, Chaigne-Delalande B, Zheng L, Pittaluga S, Matthews HF, Schmitz R, Jhavar S, Kuchen S, Kardava L, Wang W, Lamborn IT, Jing H, Raffeld M, Moir S, Fleisher TA, Staudt LM, Su HC, Lenardo MJ. Congenital B cell lymphocytosis explained by novel germline CARD11 mutations. J Exp Med 2012 209:2247-61.
Grontved L, Hager GL. Impact of chromatin structure on PR signaling: Transition from local to global analysis. Mol Cell Endocrinol. 357, 30-36.
Li M1, He Y, Dubois W, Wu X, Shi J, Huang J. Distinct Regulatory Mechanisms and Functions for p53-Activated and p53-Repressed DNA Damage Response Genes in Embryonic Stem Cells, Molecular Cell (2012)
Yang Y, Shaffer AL, Emre NCT, Ceribelli M, Wright G, Xiao W, Powell J, Platig J, Kohlhammer H, Young RM, Zhao H, Yang Y, Xu W, Balasubramanian S, Buggy JJ, Mathews LA, Shinn P, Guha R, Ferrer M, Thomas C, Staudt LM. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell 2012 21:723–737.
Koh Y, Wu X, Ferris AL, Matreyek KA, Smith SJ, Lee K, KewalRamani VN, Hughes SH, Engelman A: Differential effects of human immunodeficiency virus type 1 capsid and cellular factors nucleoporin 153 and ledgf/p75 on the efficiency and specificity of viral DNA integration. Journal of Virology. 2012.
Wang H, Jurado KA, Wu X, Shun MC, Li X, Ferris AL, Smith SJ, Patel PA, Fuchs JR, Cherepanov P, Kvaratskheila M, Hughes SH, Engelman A: Hrp2 determines the efficiency and specificity of hiv-1 integration in ledgf/p75 knockout cells but does not contribute to the antiviral activity of a potent ledgf/p75-binding site integrase inhibitor. Nucleic acids research. 2012.
Schmitz R, Young RM, Cerribeli M, Jhavar S, Xiao W, Zhang M, Wright G, Shaffer AL, Hodson D, Buras E, Lu X, Powell J, Yang Y, Xu W, Zhao H, Kohlhammer H, Rosenwald A, Kluin P, Muller-Hermelink HK, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Fisher RI , Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Pittaluga S, Wilson W, Waldmann TA, Rowe M, Mbulaiteye SM, Rickinson AB, Staudt LM. Pathogenetic mechanisms and therapeutic targets in Burkitt lymphoma from structural and functional genomics. Nature 2012 490:116-20.
Grontved L, Bandle R, John S, Baek S, Chung H-J, Liu Y, Aguilera G, Oberholtzer C, Hager GL, Levens D: Rapid genome-scale mapping of chromatin accessibility in tissue. Epigenetics Chromatin 2012 Jun 26;5(1):10.
2011
Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, Shaffer AL, Romesser P, Wright G, Powell J, Rosenwald A, Muller-Hermelink HK, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Fisher RI , Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Staudt LM. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011 470:115-119.
Martin MM, Ryan M, Kim R, Zakas AL, Fu H, Lin CM, Reinhold WC, Davis SR, Bilke S, Liu H, Doroshow JH, Reimers MA, Valenzuela MS, Pommier Y, Meltzer PS, Aladjem MI. Genome-wide depletion of replication initiation events in highly transcribed regions. Genome Research 21: 1822-1832. 2011.
CCR Sequencing Facility Presented Posters
Monika Mehta, Parimal Kumar, Vicky Chen, John Bettridge, Yongmei Zhao, Jyoti Shetty, Bao Tran. Single Cell Sequencing at CCR-Sequencing Facility. Molecular Biology in Single Cells Symposium, NCI, April 2018 & NCI Frederick Spring Research Festival, May 2018.
Keyur Talsania, Jack Chen, Tsai-wei Shen, Vicky Chen, Bao Tran, Jack Collins, Yongmei Zhao. Data Analysis for Genome Assembly and Structural Variant Detection. National Interagency Confederation for Biological Research Spring Research Festival at Fort Detrick and the National Cancer Institute, May 2018.
Vicky Chen, Tsai-wei Shen, Keyur Talsania, John Bettridge, Monika Mehta, Michael Kelly, Xiaolin Wu, Bao Tran, Jack Collins, Yongmei Zhao. High throughput Single Cell Transcriptome Sequencing Data Analysis. NIH Single Cell Symposium, April 2018.
Jack Chen, Oksana German, Sujatha Gowda, Yuliya Kriga, Christopher Hautman, Yelena Levin, Monika Mehta, Castle Raley, Jyoti Shetty, Tatyana Smirnova, Heidi Smith, Keyur Talsania, Vicky Chen, Tsai-wei Shen, Yongmei Zhao and Bao Tran. Innovative Sequencing Resources in the CCR Sequencing Facility. March 2018.
Wenming Xiao, Yongmei Zhao. A comprehensive investigation of factors impacting the accuracy of mutation detection using next-generation sequencing technology. 18-A-4219-AACR 2018.
Monika Mehta, Yongmei Zhao, Keyur Talsania, Ashley Walton, Yelena Levin, Jyoti Shetty, Elizabeth Gillanders, Bao Tran, Danielle Carrick. RNA Sequencing from Archived FFPE Tissues. AGBT Meeting, Feb 2018.
Yongmei Zhao, Keyur Talsania, Castle Raley, Monika Mehta, Jyoti Shetty, Yuliya Kriga, Sujatha Gowda, Jack Chen, Carissa Grose, Matthew Drew, Veronica Roberts, Kwong Tai Cheng, Sandra Burkett, Steffen Oeser, Robert Stephens, Daniel Soppet, Jack Collins, Bao Tran, Dominic Esposito. Draft Genome Assembly and Annotation of the Trichoplusia ni Insect Cell Line Tni-FNL. AGBT Conference 2018.
Cristobal Vera, Keyur Talsania, Ashley Walton, Sucheta Godbole, Bao Tran, Jack Collins, Yongmei Zhao. Data Analysis for Structural Variation Detection and Genome Assembly. National Interagency Confederation for Biological Research Spring Research Festival, May 2017.
Keyur Talsania, Sucheta Godbole, Ashley Walton, J. Cristobal Vera, Bao Tran, Jack Collins, Yongmei Zhao. Data Analysis Pipelines for Transcriptome Sequence Analysis. National Interagency Confederation for Biological Research Spring Research Festival, May 2017.
Monika Mehta, Yongmei Zhao, Jyoti Shetty, Castle Raley, Bao Tran. New Advancements in Next-Generation Sequencing Approaches to Address a Variety of Biological Questions. Advances in Genome Biology and Technology (AGBT) Meeting, Feb 2017.
Keyur Talsania, Sucheta Godbole, J. Cristobal Vera, Thomas Skelly, Jack Chen, Robert Stephens, Jack Collins, Bao Tran, Yongmei Zhao. Bioinformatics Support for Next-Generation Sequencing and Data Analysis at CCR-SF. National Interagency Confederation for Biological Research Spring Research Festival, May 2016.
Brenda Ho, Ashley Walton, Monika Mehta. Analysis of Illumina library preparation protocols for NGS analysis of FFPE RNA samples in cancer research. NIH Summer Intern Poster Day at NIH Bethesda campus. July 29th, 2016.
Monika Mehta, Castle Raley, Yongmei Zhao, Jyoti Shetty, Bao Tran. New Advances In Studying Cellular RNA By Next-generation Sequencing. Presented at: CCR RNA Biology Workshop at NCI Shady Grove. November 1, 2016.
CCR Sequencing Facility Presented Posters and Seminars
SF seminar Oct 28, 2020 (presentations and video recording)
Oxford Nanopore Technologies (ONT) Questions
|
Answers for General Questions
What services does the Sequencing Facility provide?
Please see the services page for a detailed list of projects we support. If your project design is not listed, please contact ccrsfhelp@mail.nih.gov or the Sequencing Facility director, to discuss the feasibility of a custom project.
Who can order services through the Sequencing Facility?
All NIH research labs are eligible to order services through the Sequencing Facility. Labs outside of CCR and NIAID will have overhead charges added.
How do I submit a sequencing request?
Please complete a sequencing proposal form at NAS request. There will be an option for Illumina, which you should select for all short read and single-cell projects, and Long Read, which you should select for all PacBio, ONT and Bionano projects. You may also contact ccrsfhelp@mail.nih.gov to discuss the available platforms and best choice for your project.
What happens after my sample is submitted?
Before sequencing, we will perform an internal QC to confirm the information in the sample manifest and notify you if any samples do not meet minimum sequencing requirements. You will then be able to choose whether to resubmit those samples or continue and sequence them at your own risks. You will be notified again when the analysis on each sample is completed and available for download.
Answers for Illumina Questions
How do I submit samples for Illumina sequencing?
Before submitting samples, ensure that the sequencing project has been discussed with the Sequencing Facility team and the NAS request submitted. Illumina service is listed under Sequencing Facility –Illumina (CCR).
You may then submit samples by delivering them at the ATRF Room D3040 (instructions for sample delivery. Be sure to include a sample manifest form with your submission as well as to send an electronic version of the form to Jyoti Shetty prior to sending your samples.
What are the requirements for submitted samples for Illumina sequencing?
Sample Quantity/Quality Requirements and Recommendations:
All samples are shipped in dry ice and the individual (1.5-2 ml) tubes are labeled clearly
| Type of library |
Minimum DNA/RNA Requirement for Library Construction |
Recommended DNA/RNA for Optimal Library Construction |
Maximum Sample Volume Requirement for Library Construction |
Additional requirements |
| ChIP DNA Sequencing |
5 ng |
10 ng |
30 µL |
Bulk of the DNA fragments in the 100-300 bp range |
| gDNA Sequencing |
100 ng |
1 µg |
30 µL |
DNA should be as intact as possible with no contamination, OD260/280 1.8–2.0 |
| mRNA Sequencing |
25 ng |
1 µg |
30 µL |
RIN should be at least 8.0, DNase treated |
| mRNA ultralow |
100 pg |
10 ng |
10 µL |
RIN should be at least 8.0, DNase treated |
| microRNA Sequencing |
100 ng |
1 µg |
6 µL |
|
| Total RNA Sequencing |
10 ng |
1 µg |
10 µL |
DNase treated, FFPE and degraded RNA can be used; DV200 < 30% not recommended |
You can use any extraction protocol as long as the DNA/RNA samples meet our sample requirements.
Here are the requirements for ATAC seq from frozen cells:
- Bulk ATAC-seq requires each sample to be present as a replicate. Triplicates are better.
- Cells for ATAC-seq should be cryopreserved at high viability. Please ensure that the cells are cryopreserved properly in freezing medium.
- Please send a minimum of 2 million cells per sample.
- Please ship the cells in dry ice.
Answers for PacBio Questions
How do I submit samples for PacBio sequencing?
Before submitting samples, ensure that the sequencing project has been discussed with the Sequencing Facility team and the NAS request submitted. PacBio service is listed under Sequencing Facility – Long Read Technology (CCR)
You may then submit samples by delivering them at the ATRF Room D3040 (instructions for sample delivery). Be sure to include a sample manifest form with your submission as well as to send an electronic version of the form to caroline.fromont@nih.gov prior to sending your samples.
What are the requirements for submitted samples for PacBio sequencing?
All samples must be sent in a 1.5 ml or 2 ml tubes.
Some requirements might be project dependent such as input of DNA if multiplexing or sequencing on multiple SMRTcells. Please contact us for more details.
Quality and quantity requirements are listed in the table below:
| Type of Library |
Minimum DNA/RNA Quantity Requirement |
Recommended DNA/RNA Quantity Requirement |
Minimum Concentration Requirement |
Quality Requirements |
| WGS |
1.5 µg |
5 µg |
n/a |
OD260/280:1.8-2.0 OD260/230:1.7-2.2 |
| Amplicons (< 5000 bp) |
200 ng |
500 ng |
n/a |
OD260/280:1.8-2.0 OD260/230:1.7-2.2 |
| Amplicons (> 5000 bp) |
300 ng |
800 ng |
n/a |
OD260/280:1.8-2.0 OD260/230:1.7-2.2 |
| HLA (Class I) |
250 ng |
1 µg |
20 ng/µL |
OD260/280:1.8-2.0 OD260/230:1.7-2.2 |
| 16S |
2 ng |
10 ng |
500 pg/µL |
OD260/280:1.8-2.0 OD260/230:1.7-2.2 |
| MAS Single Cell |
15 ng |
50 ng |
1 ng/µL |
OD260/280:1.8-2.0 OD260/230:1.7-2.2 |
| WTS |
300 ng |
1 µg |
50 ng/µL |
RIN ≥ 8.0 |
What happens during PacBio library preparation?
After initial sample QC, we proceed with library preparation. Depending on the project, the samples will be handled differently prior to PacBio library preparation. For amplicons samples, we first perform an AMPure bead clean-up that also allows us to concentrate the samples if necessary. For gDNA samples, we shear the samples to the targeted size depending on the project need and perform an AMPure bead clean-up to concentrate the samples. For WTS, we generate cDNA using polydT primers and TSO allowing us to target full length transcripts with a polyA tail. During PacBio library preparation, fragments undergo damage repair, end-repair/A-tailing and adapter ligation. The adapters are hairpin adapters and, ligated to double stranded DNA, they form a circular molecule necessary for PacBio sequencing. Barcoded hairpin adapters are also available if the project requires pooling of multiple samples. The libraries are then cleaned using AMPure beads and we perform a QC prior to setting up a sequencing run.
What are PacBio HiFi and CCS reads?
CCS stands for Circular Consensus Sequences. CCS are produced for sequencing libraries with insert size shorter than 25 kb. For CCS, the circular template (dsDNA with hairpin adapters) generated during library preparation is read multiple times and produces numerous read passes (subreads). Those subreads are then used to call a consensus sequence and generate highly accurate reads. Four passes of the molecule usually yield Q20 data while 8 passes should yield Q30 data. HiFi reads are CCS reads with > Q20.
For a quick explanation of SMRT sequencing, please watch the following PacBio video https://youtu.be/_lD8JyAbwEo
On the PacBio website: https://www.pacb.com/technology/hifi-sequencing/
What is the estimated output for PacBio sequencing?
Please note that these are estimates only as both library type and insert size are going to influence the output and it is subject to change. The Sequel II is estimated to produce 3-4.5 million raw reads. For RNA iso-seq libraries you can expect to get 3-4 million CCS reads. For WGS libraries, you can expect to get about 20-30 Gb of HiFi reads.
What can I sequence on one SMRT Cell 8M?
According to PacBio, one SMRT cell is enough to sequence a genome up to 2 Gb and a whole transcriptome, detect structural variants in up to 2 samples of ˜3 Gb genome, and multiplex numerous amplicons. For variant detection (single nucleotides, indels and structural variants) in a ˜3 Gb genome, using at least 2 SMRT cells is recommended.
See “What can you do with one SMRT cell?” for more information.
How can I extract HMW DNA?
PacBio has released a list of HMW DNA extraction protocols and QC methods that can be found at DNA preparation technical note
How can I perform target enrichment for PacBio Iso-seq?
If you are interested in long read isoform sequencing but are focused on only one or a few genes you may consider a target enrichment protocol. This protocol relies on hybridization of biotinylated probes to your cDNA target of interest and subsequent pulldown with streptavidin beads. The enriched cDNA is then amplified and prepared for PacBio sequencing. To design probes for your project please contact IDT at NGSDesign@idtdna.com or fill out a probe design request at https://go.idtdna.com/Request-consult-NGS-xGen-Custom-Hyb-Panel. To complete your sequencing request you will need to submit your probe panel in addition to your RNA samples. Please contact us for more details.
Answers for Oxford Nanopore Technologies (ONT) Questions
How do I submit samples for ONT sequencing?
Before submitting samples, ensure that the sequencing project has been discussed with the Sequencing Facility team and the NAS request submitted. ONT service is listed under Sequencing Facility – Long Read Technology (CCR)
You may then submit samples by delivering them at the ATRF Room D3040 (instructions for sample delivery). Be sure to include a sample manifest form with your submission as well as to send an electronic version of the form to Juanma Caravaca prior to sending your samples.
What happens after my sample is submitted?
Before sequencing, we will perform an internal QC to confirm the information in the sample manifest and notify you if any samples do not meet minimum sequencing requirements. You will then be able to choose whether to resubmit those samples or continue and sequence them at your own risks. You will be notified again when the analysis on each sample is completed and available for download.
What are the requirements for submitted samples for ONT sequencing?
All samples, except ONT Ultralong, must be sent in 1.5 or 2 mL tubes in dry ice. For ONT Ultralong, send the cells as a frozen pellet or cryopreserved vial in dry ice.
Quality and quantity requirements are listed in the table below:
| Type of library |
Minimum DNA/RNA Requirement for Library Construction |
Recommended DNA/RNA for Optimal Library Construction |
Maximum Sample Volume Requirement for Library Construction |
Additional requirements |
| WGS |
1 µg |
4 µg |
48 µL |
HMW DNA |
| WGS Adaptive Sampling |
2 µg |
5 µg |
48 µL |
HMW DNA |
| WGS Ultralong |
6 million human cells or the cell number equivalent to 40 µg of DNA |
n/a |
n/a |
n/a |
| Direct RNA Sequencing |
50 ng of poly(A) tailed or 500 ng total RNA |
150 ng of poly(A) tailed or 1 µg total RNA |
9 µL |
RIN ≥ 8.0 |
How can I extract HMW DNA?
We recommend the HMW Circulomics kit (NB-900-001-01) for DNA extraction.
What is the estimated output for ONT sequencing?
- GridION: up to 50 GB per flow cell
- PromethION 2 Solo: up to 200 GB per flow cell
Answers for Bionano Questions
How do I submit samples for Bionano optical mapping?
Before submitting samples, ensure that the sequencing project has been discussed with the Sequencing Facility team and the NAS request submitted. Bionano service is listed under Sequencing Facility – Long Read Technology (CCR)
You may then submit samples by delivering them at the ATRF Room D3040 (instructions for sample delivery). Be sure to include a sample manifest form with your submission as well as to send an electronic version of the form to Juanma Caravaca prior to sending your samples.
What happens after my sample is submitted?
Before sequencing, we will perform an internal QC to confirm the information in the sample manifest and notify you if any samples do not meet minimum sequencing requirements. You will then be able to choose whether to resubmit those samples or continue and sequence them at your own risks. You will be notified again when the analysis on each sample is completed and available for download.
What are the requirements for submitted samples for Bionano optical mapping?
What is the recommended coverage for Bionano optical mapping?
Answers for Single Cell Questions
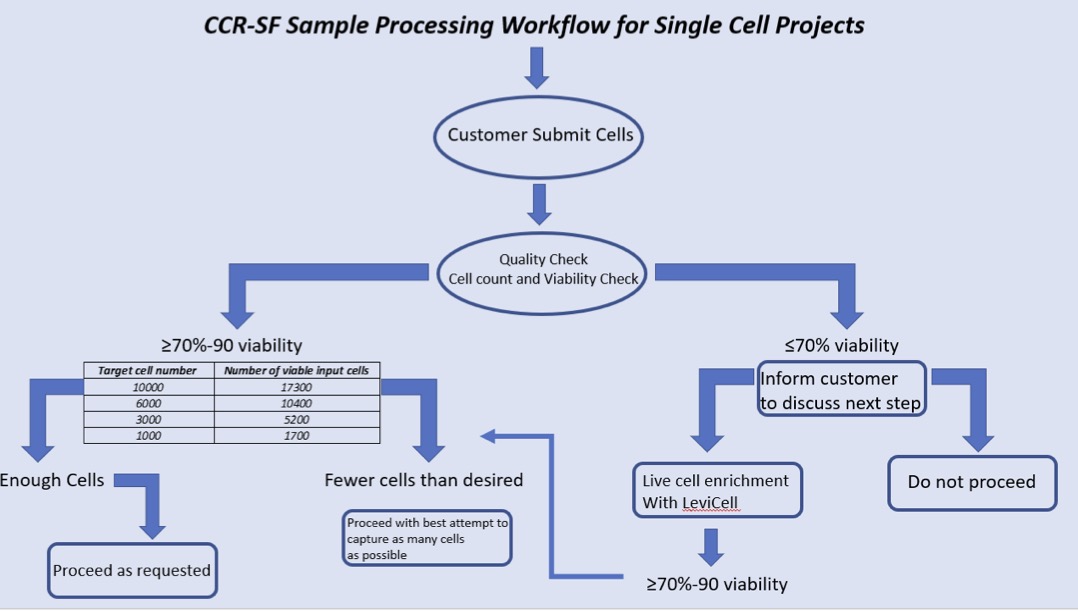
What is sample processing workflow for single cell projects?
Upon receiving single cell suspension, we check for the quality of cells and wash the cells a couple of times with PBS+BSA before loading it onto a microfluidic chip for the capture of single cells in a nanoliter size droplets along with the barcoded beads. RT takes place inside the droplet and then we break the droplets and PCR amplify the cDNA in bulk. We then purify the cDNA and check the quality. Generally, the quality of cDNA correlates very well with the final sequencing results. We then make Illumina compatible libraries from these cDNAs and sequence it on sequencer.
What is Single Cell RNA-Seq?
Single-Cell RNA-Seq provides transcriptional profiling of thousands of individual cells. This level of throughput analysis enables researchers to understand at the single-cell level what genes are expressed, in what quantities, and how they differ across thousands of cells within a heterogeneous sample.
Do dead cells impact the data quality?
10X Genomics Single Cell Protocols require suspensions of viable (90% optimal, 70-90% acceptable), single cells as input. Dead cells easily lyse, resulting in the release of ambient RNA. This cell-free RNA can contribute to the background noise of the assay and will compromise the quality of single cell data.
Clusters of dying cells typically have relatively higher levels of mitochondrial expression, lower gene counts, and more ambiguous cell type identification scores that equally or comparably match multiple major cell types.
How many cells do I need to provide?
We recommend > 1×106 cells/mL – minimum 200,000 cells/mL to load 10K live cells for non-hashing experiments and 20K-30K live cells for cell hashing experiments.
How many cells can I expect to get information for?
The capture rate of 10X is approximately 60%, depending on cell type and cell quality. When you load 10K (70-90% live) cells, you will capture around 6K cells.
How many reads do I need for my experiment?
We aim to provide 20K-50K reads/cell for gene expression libraries, 10K reads/cell for V(D)J enriched samples, 5K-10K reads/cell for CITE-Seq libraries, 50K reads/cell for single cell ATAC libraries, 20K-50K reads/cell for single cell multiome libraries as recommended by 10X genomics.
What 10X applications do you support?
We support following 10X assays: single cell gene expression (3’ and 5’), single cell immune profiling (V(D)J, TCR, BCR), Single cell multiome ATAC+Gene expression, Single Cell ATAC. We also support Mission Bio Tapestri single cell targeted DNA application.
What buffers should I use to resuspend my cells on the day of submission?
10X recommended to use 1X PBS (calcium and magnesium-free) containing 0.04% weight/volume BSA (400 μg/ml) for washing and resuspension. It is also possible to use most cell culture media with up to 10% FBS or up to 2% BSA to maintain cell health with little to no adverse downstream effects. Media should not contain excessive amounts of EDTA (> 0.1mM), or magnesium (> 3mM) as those components will inhibit the reverse transcription reaction. Any surfactants (Tween-20, etc.) should also be avoided as they may interfere with GEM generation.
How should I prepare and send my samples?
We accept fresh and cryopreserved cells. Please use 10X Genomics recommended or supported cryopreservation protocols (human/mouse) for your cells, and follow the 10X Genomics full cell preparation guide.
Fresh samples need to be at CCR_SF within 1-1.5 hour after dissociation. Fresh samples need to be loaded onto the 10X machine as soon as possible after dissociation. Please bring your samples before 2pm!
- Cell parameters: Recommended cell number: > 1×106 cells/mL. Minimum 200,000 cells/mL
- Viability: Recommended > 90%. Acceptable 70%-90%.
- Container: Please use 15mL conical Falcon tubes or 2 mL Eppendorf tubes
- Medium: You can hand your dissociated cells over in cell culture medium (up to 10% FBS or up to 2% BSA) or in 1X PBS/0.004% BSA.
- abeling: Please label tubes clearly and use permanent markers or labels. Always label your tubes on the lids and the side. Please use short unambiguous names (e.g., CTRL, IFN1).
- Temperature: Please deliver your fresh cells on ice. Please ship your cryopreserved samples on dry ice.
What is the cost per sample?
All the information about pricing is listed on our website.
What is included in the price?
We provide full service which includes administrative services, consultations, advice on experimental design, 10X genomics reagents, sample QC prior to loading, cDNA QC, post library generation QC, primary bioinformatic analysis (using the Cell Ranger pipeline).
What is the Turn Around Time for your single cell core?
It takes about 6-8 weeks from the time the sample is submitted, to data delivery after running the CellRanger pipeline. Turn around time can increase for large projects (> 48 samples) and close to the end of the fiscal year.
How should I schedule the experiment?
Please email our scientific team. They will reply you and may arrange a short meeting to discuss the project. The scheduling should be at least 2 weeks in advance. When your samples are ready, you will need to submit a NAS request listed under Sequencing Facility, Illumina (CCR). Please fill out a sample manifest form and send back to us.